Das Heidelberg Center of Human Bioinformatics HD-HuB bietet eine Cloud-Lösung für Biowissenschaftler, einschließlich umfangreicher Datensätze und standardisierter Pipelines sowie einer reinen Recheninfrastruktur, um die vielfältigen Nutzeranforderungen der biologischen Forschungsgemeinschaft zu erfüllen.
Gemeinsam verfügen die HD-HuB Partnerinstitutionen über Erfahrung in der Arbeit mit einem breiten Spektrum biologischer Datensätze und entwickeln modernste bioinformatische Methoden und Infrastruktur für deren Analyse. Unsere besonders ausgeprägte Expertise in der Verarbeitung großer menschlicher Genom- und Bildgebungsdaten (z.B. PCAWG, ICGC) beeinflusst den Hauptfokus der Heidelberg de.NBI Cloud auf Daten und Infrastruktur. Wir streben die Standardisierung von Analyseansätzen an, um die Vergleichbarkeit von Datensätzen zwischen verschiedenen Institutionen zu verbessern und integrative Analysen und Meta-Analysen zu ermöglichen. Wir sind den FAIR-Prinzipien der gemeinsamen Nutzung von Daten verpflichtet.
Das Hauptziel der de.NBI-Cloud in Heidelberg ist es, hochmoderne Tools für die Analyse von Humangenomik-, Metagenomik- und Mikroskopie-Bilddaten allgemein zugänglich zu machen.
Die Services werden in allen "klassischen" Cloud-Bereichen angeboten:
- Infrastruktur-as-a-Service (IaaS): Die Benutzer haben die volle Kontrolle über ihre virtuelle Infrastruktur. Die Ressourcen werden nicht nur in Form von Rechenleistung über virtuelle Maschinen (VMs) bereitgestellt, sondern umfassen auch Netzwerke, Firewalls und Speicher. Zusätzlich bieten wir der Community spezialisierte Hardware (z.B. GPUs, FPGAs und High-Memory-Knoten) für modernes paralleles wissenschaftliches Rechnen in großem Maßstab (z.B. Matrixfaktorisierung, Deep Learning). Dies gibt den Benutzern die Flexibilität, die sie benötigen, um die Hardware an ihre Anforderungen anzupassen und Workflows und Pipelines effizient zu entwickeln und gleichzeitig die Leistung zu optimieren.
- Plattform-as-a-Service (PaaS): Den Benutzern wird eine "Plattform" zur Verfügung gestellt, die es ihnen ermöglicht, ihre wissenschaftlichen Aufgaben zu erfüllen. Diese werden in der Regel in Form von Workflow-Frameworks wie Galaxy https://github.com/BMCV/galaxy-image-analysis, Roddy, Butler (mit Multi-Cloud-Bereitstellung, Konfigurationsmanagement, umfassender Überwachung und Anomalieerkennung sowie vorgefertigten Pipelines für Genom-Alignment, Keimbahn- und somatisches Varianten-Calling und R-Skript-Ausführung) oder den ICGC-Pipelines bereitgestellt, sind aber nicht darauf beschränkt. Wir möchten mehrere VM-Images bereitstellen, jeweils mit einem kuratierten Satz von Software, der sich auf einen bestimmten Bereich der Bioinformatik konzentriert, um Forschern der Biowissenschaften einen schnellen Einstieg zu ermöglichen.
- Software als Dienstleistung (SaaS): Den Nutzern werden vollständig etablierte Softwarepakete oder Workflow-Systeme zur Verfügung gestellt, die sie direkt nutzen und auf ihre Probleme und Daten anwenden können. Dies eignet sich ideal für explorative Analysen von Genomik und Metagenomik, Bildgebungsdaten und darüber hinaus. Darüber hinaus stellen wir maßgeschneiderte Software und Workflows im Rahmen der Weitergabe unseres Fachwissens an Kooperationspartner innerhalb der de.NBI-Community zur Verfügung. Es ist auch eine großartige Ressource für angehende Bioinformatiker und zur Unterstützung der Lehre.
- Daten als Service (DaaS): Benutzern wird der Zugang zu großen Datenpools ermöglicht, die für die biowissenschaftliche Forschung unerlässlich sind. Diese stellen eine äußerst wertvolle Ressource dar, können aber aufgrund der Größe oder der Zugriffsregelungen für die Daten oft nicht an das Unternehmen des Nutzers übertragen oder dort gespeichert werden. Indem die de.NBI-Cloud den Zugang zu diesen Daten ermöglicht, wird sie die Entwicklung neuer Bioinformatik-Methoden vorantreiben und das Benchmarking von Analysetechniken an zuvor unzugänglichen Datensätzen erleichtern. Je nach Bedarf können Benutzer autorisierten Zugriff auf eingeschränkte Datensätze erhalten, wie z.B. Referenzdatensätze der EBI-ENA oder allgemeine Datenquellen wie dbGaP und GENCODE. Wir haben begonnen, uns dem neuen ICGC-Cloud-Computing-Projekt für kollaborative Forschung anzuschließen. Die Rohdaten und die interpretierten Daten, die aus den Sequenzen von Tumoren und passenden Normalgeweben verarbeitet werden, werden von der Heidelberg de.NBI Cloud zur Verfügung gestellt, um analytische Ergebnisse zu generieren und neue Tools zu entwickeln.
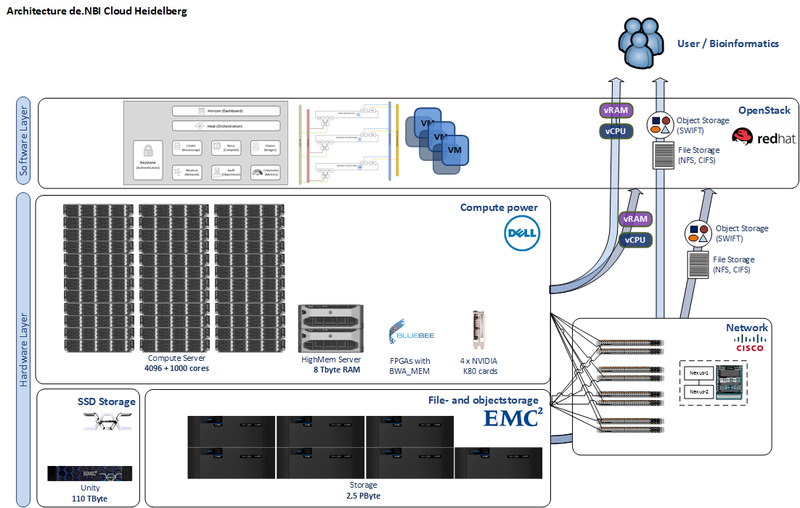
Die de.NBI Cloud-Infrastruktur in Heidelberg besteht aus 4096 Kernen, 32 TByte RAM, 8 TByte RAM in High-Memory-Knoten, 100 TByte SSD und 2,5 PByte Speicherkapazität. FPGAs sorgen für eine schnelle Verarbeitung von genomischen Alignments. GPUs stehen für Deep-Learning-Ansätze auf riesigen Mengen von markierten Daten zur Verfügung.